Tìm hiểu về căn bệnh hiếm gặp mang tên: Hội chứng Apert
Hội chứng Apert hay còn gọi là bệnh xương cứng sớm cục bộ, và đường thở hẹp là một căn bệnh hiếm gặp nhưng rất quái ác và gây nhiều bệnh nặng cho trẻ em.
Hội chứng này xuất hiện là do sự biến đổi trong tinh dịch. Sự biến chuyển này ngăn chặn sự phát triển của đầu óc, khiến các ngón chân và ngón tay dính liền vào nhau và kích thích sự phân chia chuỗi tế bào hình thành nên tinh dịch.
Cứ 70.000 trẻ thì có 1 trẻ bị mắc chứng Apert bẩm sinh. Sự thay đổi ADN xuất hiện trên một gene đơn lẻ trên nhiễm sắc thể 10 của đứa bé và có liên quan mật thiết đến tuổi của người cha. Hội chứng này khiến cho cơ thể đứa bé còi cọc và dẫn đến tình trạng thiếu phát triển sụn, theo đó các cơ quan hoặc bộ phận cơ thể mềm vẫn phát triển bình thường nhưng xương thì không hề phát triển.
Nguyên nhân của Hội chứng Apert
Hội chứng Apert được gây ra bởi một đột biến hiếm hoi trên một gene duy nhất. Gene đột biến này bình thường chịu trách nhiệm để các xương khớp với nhau vào đúng thời điểm trong quá trình phát triển.
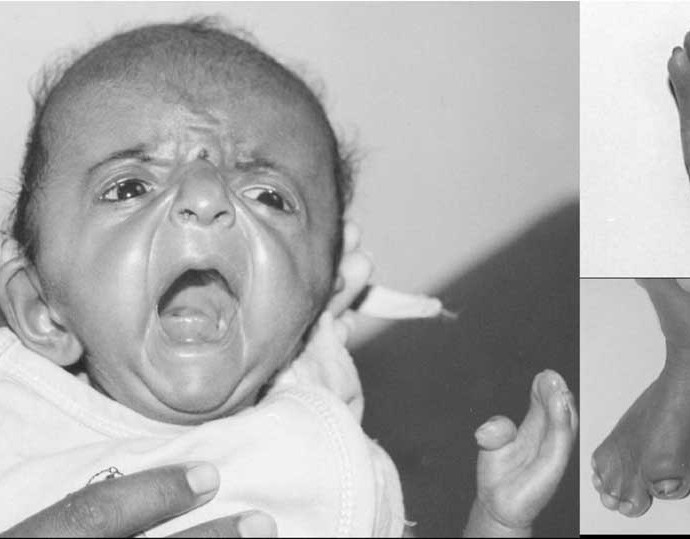
Một bé mắc hội chứng Apert hay còn gọi là bệnh xương cứng sớm cục bộ.
Hội chứng Apert là rối loạn NST thường, khoảng hai phần ba các trường hợp là do một đột biến C thành G ở vị trí 755 trong gene FGFR2, gây ra thay đổi từ Ser thành TRP trong protein.
Trong hầu hết các trường hợp, hội chứng Apert gene đột biến có vẻ như là ngẫu nhiên. Chỉ có khoảng một trong 65.000 em bé được sinh ra với hội chứng Apert. Hội chứng Apert có thể di truyền từ người lớn mắc bệnh với xác suất 50%, hoặc có thể do đột biến tự phát.
Triệu chứng Hội chứng Apert
Gen khiếm khuyết ở trẻ sơ sinh với hội chứng Apert khiến các xương sọ bị chèn ép bởi hộp sọ đóng kín sớm, quá trình này gọi là craniosynostosis. Não tiếp tục phát triển bên trong hộp sọ nhỏ hẹp bất thường, gây áp lực lên xương hộp sọ và khuôn mặt. Hộp sọ bất thường và tăng trưởng khuôn mặt ở hội chứng Apert tạo ra các dấu hiệu và triệu chứng chủ yếu sau:
- Sọ hình tháp do dính khớp sọ sớm.
- Mắt trũng, thường với mí mắt kém đóng, cơ chuyển động mắt mất cân đối.
- Một khuôn mặt giữa trũng. Kém phát triển phần giữa mặt dẫn đến xương gò má thấp và lồi mắt.
Các triệu chứng khác cũng là kết quả từ sự tăng trưởng bất thường của sọ:
- Có thể có kém phát triển trí tuệ (trong hầu hết các trẻ em bị hội chứng Apert).
- Tắc nghẽn gây ngưng thở khi ngủ.
- Khiếm thính do nhiễm khuẩn tai, viêm xoang, nghe kém.
- Dính xương của các ngón tay và chân (syndactyly) – với hai bàn tay hoặc bàn chân. Một số trẻ em bị hội chứng Apert cũng có dị dạng về tim, tiêu hóa, hoặc các vấn đề hệ thống tiết niệu mà không gặp ở các hội chứng dính sớm khớp sọ khác.
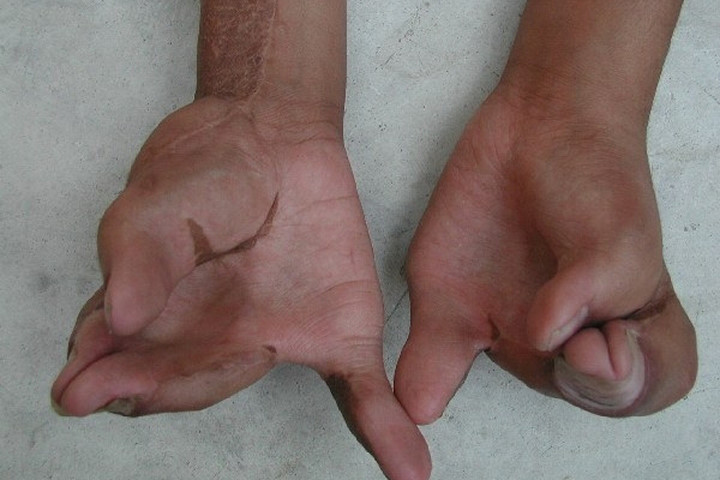
Các ngón tay bị dính vào nhau và co quắp.
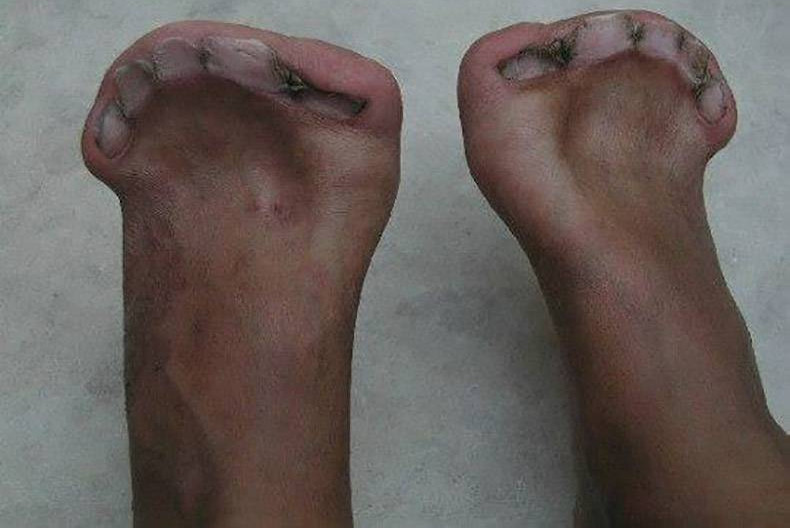
Bàn chân dị dạng với 5 móng chân nhưng hoàn toàn không có ngón.
Chẩn đoán Hội chứng Apert
Các bác sĩ thường nghi ngờ hội chứng Apert hoặc hội chứng craniosynostosis sơ sinh khác vì vẻ bề ngoài của trẻ sơ sinh. Thử nghiệm di truyền thường có thể xác định hội chứng Apert hoặc nguyên nhân khác của sự hình thành hộp sọ bất thường.
Điều trị Hội chứng Apert
Trẻ mắc hội chứng Apert có thể phát âm bất thường. Thường không có âm mũi do phần giữa mặt kém phát triển, mũi nhỏ, và vòm miệng mềm rất dài. Nếu có khe hở vòm miệng, bệnh nhân mắc hội chứng Apert cũng có thể phát âm không có âm mũi. Phát âm thường không rõ do khớp cắn lệch và vòm miệng nhô cao. Nghe kém hoặc chậm phát triển toàn thể cũng có thể ảnh hưởng đến phát triển ngôn ngữ và khả năng phát âm. Phẫu thuật để sửa chữa các kết nối bất thường giữa xương là phương pháp điều trị chính cho hội chứng Apert.
Thời điểm tốt nhất để giải phóng các đường khớp sọ dính khoảng từ ba đến sáu tháng tuổi; có thể thực hiện tới 18 tháng tuổi. Phẫu thuật sớm cho phép não trẻ có không gian để phát triển. Ngoài việc giải phóng các đường khớp sọ, xương trán biến dạng và các xương khác sẽ được tái định vị để sửa chữa lồi mắt và dị dạng phần trên của mặt. Bác sỹ phẫu thuật thẩm mỹ và bác sỹ ngoại thần kinh sẽ kết hợp mổ để đem lại hiệu quả cao nhất cho cuộc mổ. Nếu phần giữa mặt và phần trên hàm không phát triển đúng mức thì nên phẫu thuật tiếp khi trẻ lớn. Dính tay thường được tách trong những năm đầu đời để tay thẳng hơn và chức năng tốt hơn. Đôi khi, có thể phải phẫu thuật nhiều lần để trẻ có đầy đủ chức năng hoạt động.
Phẫu thuật cho hội chứng Apert diễn ra trong ba bước:
- Giải phóng xương sọ dính sớm (craniosynostosis). Một bác sĩ phẫu thuật tách xương hộp sọ dính bất thường và một phần sắp xếp lại một số xương trong số đó. Phẫu thuật này thường được thực hiện khi một đứa trẻ từ 6 đến 8 tháng tuổi.
- Phẫu thuật tịnh tiến phần giữa mặt (Midface advancement). Khi đứa trẻphát triển với hội chứng Apert, xương mặt trở thành lệch. Bác sĩ phẫu thuật cắt xương hàm và má và đặt lại vị trí bình thường cho các xương này. Phẫu thuật có thể được thực hiện tại bất kỳ thời gian nào từ 4 đến 12 năm tuổi. Phẫu thuật chỉnh hình có thể được bổ sung cần thiết, đặc biệt là khi midface advancement được thực hiện sớm khi trẻ còn nhỏ.
- Sửa chữa đôi mắt rộng (hypertelorism). Bác sĩ phẫu thuật sẽ tiến hành loại bỏ một nêm của xương trong hộp sọ giữa hai mắt, đưa hốc mắt lại gần nhau hơn, có thể điều chỉnh hàm kết hợp với phẫu thuật này.
Đời sống
-

Những xác ướp bí ẩn ở thị trấn Colombia
-

Hạt Quinoa là gì? Lợi ích vàng của hạt quinoa đối với sức khỏe
-

Kỳ lạ cô gái 18 tuổi ở Trung Quốc mắc bệnh "luyến ái não"
-

Tiểu hành tinh quay nhanh kỷ lục đâm xuống Trái Đất
-

Vì sao Trái đất sẽ không bao giờ bị "nuốt chửng" nếu một ngày nào đó Mặt trời hóa lỗ đen?
-

Hố xanh khổng lồ được phát hiện: Con người chưa đủ khả năng khám phá!
Sức khỏe
-

Những kỹ năng giúp trẻ tránh bị xâm hại tình dục
-
Ăn cá có thể giúp người già tránh bệnh Alzheimer
-
Úc: cấy ghép tế bào tạo insulin cho bệnh nhân tiểu đường type I
-
Tạo ra phân tử nhân tạo chống lại vi khuẩn kháng thuốc
-
Thai suy dinh dưỡng khó chẩn đoán
-
Mang thai trong khi đang... có bầu



Tiêu điểm
-
Việt Nam có 1 loại “nấm trường thọ” chứa hơn 400 dưỡng chất quý giá
-
Việt Nam có một loại củ phơi khô được ví như "thuốc trường thọ"
-
Sai lầm khi ăn dưa chuột khiến dinh dưỡng hao hụt, lợi ích sụt giảm: Hóa ra nhiều người vẫn thường làm
-
Một loại củ ngọt mát tốt ngang nhân sâm tổ yến rất sẵn ở Việt Nam
-
Đã đến lúc thế giới nên ngủ trưa
-
Loại lá vị tanh nồng là "thuốc hạ đường huyết" cực nhạy, mọc đầy ở bờ ruộng của người Việt
-
Giấc ngủ chia con người thành 4 kiểu như thế nào?







-

Công nghệ mới
-

Phần mềm hữu ích
-

Khoa học máy tính
-

Phát minh khoa học
-

AI - Trí tuệ nhân tạo
-

Khám phá khoa học
-

Sinh vật học
-

Khảo cổ học
-

Đại dương học
-

Thế giới động vật
-

Danh nhân thế giới
-

Khoa học vũ trụ
-

1001 bí ẩn
-

Ngày tận thế
-

Chinh phục sao Hỏa
-

Kỳ quan thế giới
-

Người ngoài hành tinh - UFO
-

Trắc nghiệm Khoa học
-

Lịch sử
-

Khoa học quân sự
-

Tại sao
-

Địa danh nổi tiếng
-

Bệnh và thông tin bệnh
-

Y học - Sức khỏe
-

Môi trường
-

Bệnh Ung thư
-

Virus Covid 19
-

Ứng dụng khoa học
-

Khoa học & Bạn đọc
-

Câu chuyện khoa học
-

Công trình khoa học
-

Sự kiện Khoa học
-

Thư viện ảnh
-

Góc hài hước
-

Video